Vanessa Sánchez
LA PATRIA | Manizales
Mu-co-po-li-sa-ca-ri-do-sis, sí, aunque esta palabra cuesta pronunciarla, se trata de una enfermedad rara, hereditaria y genética, en la que los pacientes tienen estatura baja, tronco corto y abdomen abultado, por el crecimiento anormal del hígado y el bazo.
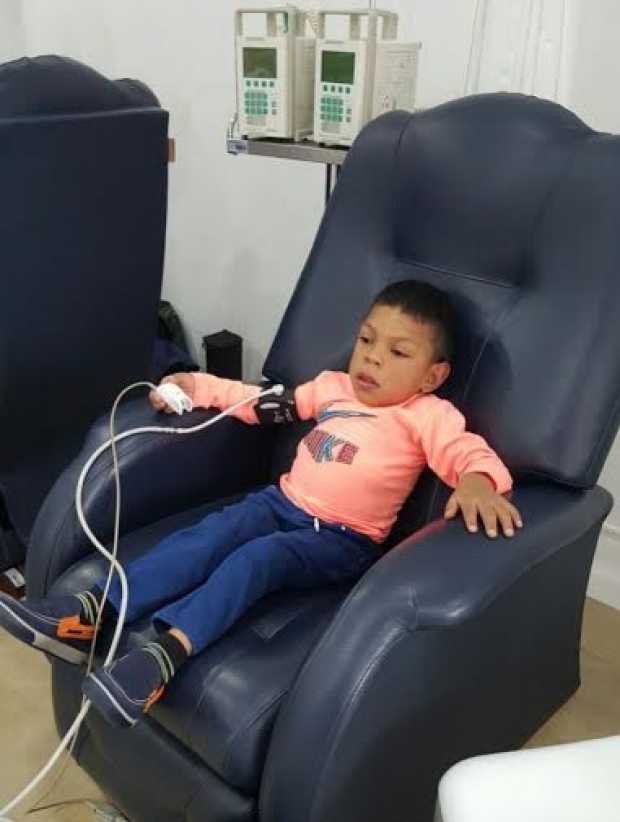
El claro ejemplo del padecimiento lo tiene Alejandro Arenas, a quien le diagnosticaron en el 2009 la enfermedad. Paula, su mamá, notó que su crecimiento se detuvo después de cumplir 18 meses. El menor bajó tanto de peso que se le empezaron a notar las malformaciones en sus huesos.
La familia vivía en el Páramo de Letras, a hora y media de la carretera principal. Viajar a Manizales a la espera de un diagnóstico fue un tráuma. "Caminaba un trayecto muy largo con el niño en brazos, porque a él le dolía mucho montar a caballo. En ese momento estábamos afiliados a Caprecom y estuvimos en muchas citas sin saber qué tenía. Apenas a los 5 años nos dijeron que padecía mucopolisacaridosis tipo 4 (morquio), es decir, en la que no había compromiso cognitivo".
Aunque la noticia la preocupó, fue más traumático el acceso al tratamiento. El genetista le ordenó unos medicamentos que la EPS nunca le entregó, ni por tutela ni con desacatos. Ante la situación, Paula viajó a Bogotá a buscar una alternativa para su hijo.
"Conseguí un empleo en un cultivo de flores y me afiliaron a Famisanar. Empezamos de cero y desde febrero del año pasado pudo recibir los medicamentos. Debido a que la enfermedad avanzó, ahora tiene un soplo en el corazón, una afección respiratoria aguda, porque su caja torácica es de un niño de 2 años con pulmones de uno de 8", agrega.
El sueño de Alejandro es estudiar como su hermano, de 5 años, que lo supera en tamaño, pero no en conocimiento. Aunque la enfermedad no tiene cura y con el paso del tiempo los síntomas se agudizan, recibe cada ocho días unas terapias especiales para ofrecerle una mejor calidad de vida.
Trabas
Luz Victoria Salazar, directora de la Asociación Colombiana de Pacientes con Enfermedades de Depósito Lisosomal (Acopel), señala que 1 de cada 100 mil habitantes padece de mucopolisacaridosis, y debido a que se considera como una afección rara, la primera dificultad que encuentran en el sistema de salud es un diagnóstico a tiempo. De tres a 10 años se tardan estos pacientes en ser diagnosticados.
"Cuando por fin dan con el dictamen ya presentan síntomas irreversibles y la recuperación no es la misma. Debido a que el médico determina un tratamiento que es producido para pocas personas, entonces es muy costoso y de difícil acceso", indica.
Salazar comenta que casi todos los procesos para acceder a las terapias y la medicación acaba por vía jurídica y a veces ni con tutela logran el cometido, pues pasado un mes las EPS interrumpen los tratamientos y los pacientes pierden el camino que han recorrido. En Colombia, se calcula que hay aproximadamente 440 personas con esta enfermedad, de las cuales solamente hay detectados 250 casos, principalmente niños.
"Desde la entidad ofrecemos medios para que las personas encuentren el diagnóstico, información sobre la enfermedad, y apoyo psicosocial. Abogamos porque dentro de las políticas públicas se incluyan las enfermedades huérfanas y el sistema de salud los tenga en cuenta como lo contempla la Ley Estatutaria", concluye la directora.
Rara y nueva
El neuropediatra Jhon Jairo Silvestre señala que esta es una enfermedad de origen genético, es decir, el paciente nace con una condición que se va expresando a lo largo de la edad. Su causa es la ausencia o el mal funcionamiento de una enzima, necesaria para desechar sustancias relacionadas con la formación y sostén de los tejidos. Su acumulación altera órganos vitales como corazón, pulmones y afecta el desarrollo físico y mental.
Cuenta que normalmente el hígado y el bazo aumentan de tamaño, hay deformidades óseas, opacidad en la córnea y hernias e infecciones repetitivas.
"La expectativa de vida depende de la severidad del cuadro, porque hay unas formas muy agudas en las que las personas viven hasta los 20 años máximo. No obstante, no para todos los tipos de mucopolisacaridosis existe tratamiento, puntualmente en la tipo 4 hasta hace dos años se desarrolló una terapia de medicamentos que antes no existía", explica.
Silvestre señala que en Manizales atendió dos casos y uno de ellos falleció a sus 20 años. Indica que en Caldas se presume que deben haber más casos porque el departamento se caracteriza por las relaciones endogámicas, es decir, que los padres de los niños son primos o tíos y en este tipo de familias es posible que la coincidencia en los genes provoque la alteración genética.
EL DATO
Dentro de la enfermedad hay varias clasificaciones. La tipo 1 y 2 afecta la parte cerebral y los pacientes muestran retardos mentales. La tipo 3 y 4 afecta el sistema óseo.
El uso de este sitio web implica la aceptación de los Términos y Condiciones y Políticas de privacidad de LA PATRIA S.A.
Todos los Derechos Reservados D.R.A. Prohibida su reproducción total o parcial, así como su traducción a cualquier idioma sin la autorización escrita de su titular. Reproduction in whole or in part, or translation without written permission is prohibited. All rights reserved 2015


